Antibody discovery is becoming more data driven
I’m sure many people have heard phrases like “big data is going to change drug discovery”, or “deep learning is going to revolutionize the search for new medicines”. But what does this look like in practice when it comes to antibody discovery? In this post, we’re going to talk about how antibody discovery is becoming more data driven.
The status-quo
To understand how data is changing antibody discovery, it is probably helpful to get an overview of the current approaches to antibody discovery so we can appreciate how data and algorithms are being used in novel ways. In this section, I will provide that background.
Typically, in an antibody discovery campaign. You are trying to obtain antibodies that are optimal across several different dimensions. For example, you may want antibodies that bind to orthologs of your target protein so that you can test out the safety and efficacy of your antibody in preclinical trials using model organisms. You often (although not always) want your antibodies to have high affinity for their target. It is also important that your antibodies are specific for their target and do not have any off-target effects (through their interaction with proteins other than their intended target).
Strategies exist to enrich for these properties, most of which revolve around manipulating the parameters of your biopanning/selection process. I briefly explained how selections worked in my first blog post which you can find here, but I will go into more detail in this post.
Any antibody discovery campaign will start with an antigen i.e. your drug target - typically a protein, or protein complex which acts as molecular bait to capture binding antibodies from your antibody library. As with bait you need some way to recover the antigen along with its bound antibody. This is typically done by conjugating your antigen to biotin molecules. Biotin is a small molecule that has the useful property of binding with extremely high affinity to another protein called streptavidin (or its close relative neutravidin). Streptavidin can be attached to tiny magnetic beads which can be pulled out of solution with a magnet, allowing you to recover the biotin conjugated antigen along with any antibodies bound to it.
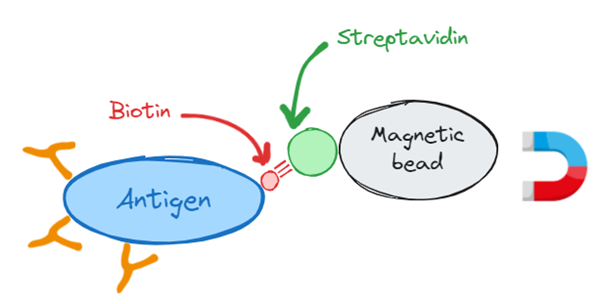
To summarise, we conjugate biotin to our antigen, we add biotinylated antigen to our antibody library and allow specific antibodies to bind, we then add streptavidin coated magnetic beads to pull out the antigen and it’s bound antibody. This is the selection process in a nutshell. Some details have been missed out here. For example, the antibodies are typically expressed on phage particles that contain the DNA encoding the antibody, however it is good enough for our purposes.
So, now that we have a handle on the basic selections process, let’s go back to our initial question; how can we manipulate the process to ensure the antibodies we get out fulfil the various criteria that are important to us (e.g. affinity, specificity, cross reactivity etc)? At a high level, the antibody selection process is divided up into a series of “arms” or “strategies”. Within each arm we calibrate the selection pressures we apply to our antibody libraries in the hope that we will be enriching the population we get out with the properties we are interested in.
To illustrate I have included a diagram that represents the course an actual antibody discovery campaign might take. This campaign is split into three arms, each arm having 4 rounds. Looking at the first arm (far left) as an example we see that we start off our selections with human antigen at a concentration of 100 nanomolar. We take the output of this selection and use it as input to another selection, however in this selection we use the mouse ortholog of our antigen. Again, the output from this round is used as input for round three where we drop the concentration of our antigen to 10 Nanomolar. By swapping the species ortholog of our antigen and dropping the concentration, we are changing our selection pressure in order to promote cross reactivity and affinity (lower antigen concentrations increase competition for epitopes and stronger binders should win out over weaker ones).
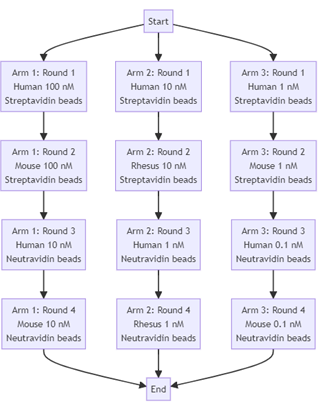
If we look at the other arms, we see that we are applying different selection pressures (selecting for rhesus cross reactivity instead of mouse for example). At the end of the process outputs from the different arms and rounds will be sampled and screened in binding and functional assays. This example campaign is again a simplification, there would likely be more strands than those I’ve shown here, however the point is that we are manipulating experimental parameters to try to guide the selection process to give us the output we want. For programmers in the audience, you can think of this as if we are applying a pipeline of filters to our input.
As is probably obvious from the diagram this approach is quite complex! In fact, it’s more complicated than the diagram might imply, as it does not include deselections (effectively an anti-selection to remove off target binding, where instead of using the output - our off-target binders - for the next round we throw it in the bin and use what’s left instead). As you can imagine, configuring an antibody selection campaign requires careful thought and expertise. How much should we decrement the antigen concentration by each round? At what point should we do deselections? How many rounds should we do for each arm before we screen? These are sometimes difficult questions, however getting them right is key to a successful campaign. For example, if you set the antigen concentration too low, the selection pressure will be too high and the result is often a collapse in diversity of your library, while setting it too high means your selection pressure will be too weak and you will fail to enrich your output for target binding antibodies.
In addition to being tricky to set up, these approaches require multiple sequential rounds of selection so that we can manipulate experimental parameters to modify our selection pressure. Unfortunately, fundamental dynamics of population change in our antibody library make this difficult. In between each selection round, our outputs antibody population is amplified in E. coli. Within our antibody population there will be variation in replication rates (for example some antibody sequences may be shorter or be more amenable to synthesis by the bacterial replication machinery). A small growth advantage, compounded over many rounds of amplification, often results in some antibodies becoming dominant in the population. This phenomenon is undesirable as it reduces the diversity of our output, and these dominant clones are often weak binders or even non binders (they may have survived the selections due to their interactions with plastic or magnetic beads). Some of the evidence for this is laid out nicely in this paper, where they show that no matter the size or the complexity of the antigen being selected against, diversity seems to drop at about the same rate as selection rounds progress.
So in summary, antibody selection campaigns are difficult to set up, require multiple arms to hedge against failure, And necessitate multiple sequential rounds to enable the application of different selection pressures, risking diversity collapse due to growth advantage effects. In addition to these drawbacks, there are many important properties that are not typically selected for during the initial campaign such as expression yield, stability or reduced immunogenicity. The advantage of this approach however is that it does not require any understanding of the relationship between antibodies and the properties we care about. Our selection process is a black box. We input our antibody library, and the output is a subpopulation enriched for our property of interest (we hope). However new techniques for gathering high quality data coupled with powerful new algorithms to derive insight from this data, may enable us to understand these relationships and redesign the discovery process to give us a better chance of success.
Data-driven antibody discovery
To understand how data can impact antibody discovery we have to understand a simple fact. Antibody properties are determined by their 3-dimensional structure, which is in turn determined by their 1-dimensional sequence amino acid sequences. For example, binding affinity of an antibody for its antigen is determined by shape and chemical complementarity between the antibody binding loops and their epitope (binding region) on the antigen, much like a lock and key. If we can learn the complex relationship between 1-dimensional linear sequence and 3-dimensional structure and chemical properties, then we can learn the relationship between antibody sequences and the properties we are interested in. With this information, we can predict which antibodies are likely to be fit for purpose, before doing the experiment, saving time and money, or even design then from scratch. I mentioned that algorithms and data are central to the shift we are discussing. Lets talk about these algorithms.
The class of algorithms I am talking about are deep learning algorithms or neural networks. These networks are loosely modelled on the way the human brain works. A detailed account of how neural networks work is outside the scope of this article, however broadly they can be used to approximate arbitrarily complex functions that map from some input space to some output space. For example, the space of human text descriptions to the space of 2D images, as is done by the likes of DALL-E and Midjourney.
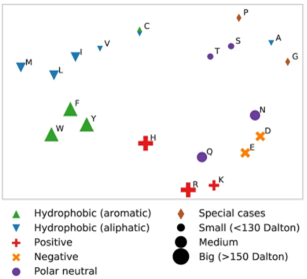
The plot on the left is a visualization of the intermediate output of one of such model, that was trained specifically on amino acid sequences .The visualization shows one view of the models internal representation of amino acids. It is clear from the plot that the model has learned some key biophysical properties of amino acids such as their size, charge and hydrophobicity (as it places amino acids with similar properties near each other in 2D space). What is fascinating about this, is that this model was never directly trained on biophysical properties of amino acids. It was simply trained to infill masked residues in the sequence. Despite the simplicity of the training task, the model was nonetheless able to learn some core features of protein sequences.
So deep learning models trained on amino acid sequences of proteins can learn complex biological patterns in sequence data if they are relevant to their training task. An example of the application of these techniques to antibody discovery came from an academic group led by Sai Reddy. In this work, the group wanted to optimize the therapeutic antibody Trastuzumab. They generated a relatively small library of trastuzumab (representing less than 0.01% of the possible sequence space), and sorted them for antigen binding using FACS. The resulting populations were positive and negative binding sequence populations were labelled, and a classification model was trained to predict binding to. This allowed the team to rank and filter an in-silico library for target binding that was much larger (3 orders of magnitude) than the one used for training. Building and screening a library of this size directly would have been costly and time consuming. In addition to being able to screen a much larger library in silico, they were able to rank and filter on a number of other predicted biophysical properties including low viscosity in solution and good solubility. This approach therefore opens the door to the kind of multiparameter optimization that we saw was difficult to do with traditional selection approaches.
Another important property of antibody therapeutics that we have touched on is low immunogenicity. Exogenously delivered antibody therapeutics are foreign proteins after all, and our bodies tend to try to remove foreign entities if they are detected. Many of the early antibody therapeutics brought to market were derived from mice and had subtly different features from human antibodies. In many cases this meant that after the first dose, these antibodies would become useless, as the body would have developed its own antibodies to bind to it and remove it from the system as soon as it was injected. Several different approaches have been developed, both experimental and in silico, to predict likely immunogenicity in antibodies, either so they can be fixed, or abandoned early. Relevant to this post are new techniques that leverage both deep learning and public sequence repositories to learn the sequences features that distinguish human from non-human antibodies.
One such approach is implemented by BioPhi, a platform for assessing immunogenicity in antibodies. BioPhi is made up of two components, but one of them (Sapiens) relies on exactly the kind of deep learning models I introduced earlier. The data to train these models came from the Observed Antibody Space (OAS) maintained by the excellent folks at the Oxford Protein Informatics Group (OPIG). This database contains millions of human and non-human sequences, enabling the development of Sapiens which was trained to predicted species labels for antibody variable domains. With this model, putative therapeutics can be scored on their relative ‘humanness.’ Indeed, using the scores derived from this model, many of the same modifications originally proposed in humanized antibodies currently in the clinic were recapitulated, despite the fact that those modifications had often been arrived at via expensive trial and error experimentation and with the input of very specialized human knowledge.
Those were just a couple of examples; however I hope it illustrates that with new algorithms and better data, we are getting to the point where we are able to accurately and reliably predict antibody properties. The upside of this is huge. The discovery process could become faster and cheaper, an important goal, as the costs of drug discovery more generally have increased drastically in recent decades, and could increase the safety and efficacy of therapeutics. Several barriers stand in the way of this becoming reality however. A key requirement of deep learning is data, and typically lots of it. While this has not typically been a problem in mainstream deep learning that relies on plentiful data scraped from the web, the story in biology is different. Generating data is often expensive and time consuming. So much so that in many cases it outweighs the benefits of a data driven approach in the first place. This difficulty is compounded by the fact that datasets of any meaningful size are siloed within companies, and even within companies are often not standardized enough to be used.
For the future of antibody discovery to become the present, the cost and time it takes to generate data have to come down. This is slowly happening, with innovations in sequencing technology rapidly driving down the cost of generating sequence data. On the algorithmic side, approaches that reduce the data requirements for effective learning will be key, such as more effective ways of training models that leverage larger more readily available data formats . Another trend that could shift the needle would be if companies and academic groups get more comfortable collaborating and sharing datasets (although with the level of secrecy in the industry I wouldn’t hold my breath on this one). Finally, it will require those that currently work in the field to understand the potential of these new technologies and get behind them. I hope this post contributes in a small way to that.